CHAPTER 19
Sleep-Wake Disorders
In this chapter we review the current status of diagnosis and treatment of sleep-wake disorders for our target audience: clinicians in general psychiatric practice who are not specifically trained in sleep disorders medicine but who will nonetheless encounter sleep complaints in many (if not most) of their patients. We cannot be comprehensive in our coverage of sleep disorders: the current second edition of the International Classification of Sleep Disorders (American Academy of Sleep Medicine 2005), considered by many to be the definitive classification for sleep disorders, lists over 80 specific diagnoses in eight general categories. Instead, our intention is to provide sufficient information and guidelines so that the clinician is unlikely to miss major sleep disorders and their detrimental consequences.
This chapter is divided into three main sections: the first provides a summary of the basic science of wakefulness and sleep, the second introduces and briefly describes the various sleep-wake disorders included in DSM-5 (American Psychiatric Association 2013) and the changes from the previous edition of DSM, and the third presents a four-step evaluation to guide the clinical approach to a sleep complaint.
Borbely and Achermann (1999) have postulated a clinically useful two-process model for sleep and sleep regulation: Process S is the Sleep homeostatic drive, which increases in strength as a function of time awake. The opposing Process C is the Circadian arousal drive, which increases during the day, thus maintaining the state of wakefulness and preventing Process S from taking over until the normal bedtime approaches, at which point the Process C arousal drive begins to diminish and Process S is permitted to take over. Sleep ensues, with its reversal of the neurometabolic effects of the waking state. Most sleep disorders can be conceptualized as a disorder of Process C, Process S, or both.
Two fundamental issues underlie sleep and wakefulness. The first is the fact that the brain can normally be in only one of three possible states: 1) wakefulness, 2) non-rapid eye movement (NREM) (slow wave) sleep, or 3) rapid eye movement (REM) sleep. There are control systems (neurophysiological switches) that regulate shifting from one state to another. Impaired control of brain states is dysfunctional and contributes to several sleep disorders, including narcolepsy (abnormal shifting between wakefulness, NREM sleep, and REM sleep) and parasomnias such as sleepwalking (admixture of sensorimotor elements of wakefulness with NREM sleep). The second fundamental issue is that the probability of being in one of the three different states is controlled by the circadian timing system (Process C), disruptions of which contribute to several common sleep disorders.
Our discussion of the wake-sleep control systems will begin with the circadian timing system, knowledge of which is essential to understanding sleep complaints. We will then discuss the wake and sleep control systems in turn.
The timing of sleep onset is largely controlled by a person's circadian arousal. drive (Process C), closely associated with body temperature, which increases during the day and early evening, maintaining wakefulness even though the homeostatic sleep drive (Process S) is increasing with time awake, and which begins to decrease around the time of normal sleep onset, thus permitting sleep. Circadian rhythms are controlled by the suprachiasmatic nucleus (SCN) of the anterior hypothalamus, and the circadian clock controls many internal rhythms, including the sleep-wake cycle, which it keeps in tune with the day-night pattern of the external environment. The circadian clock also maintains temporal organization of internal physiological processes and makes sure that their changes are coordinated with one another. Circadian rhythms are genetically determined and persist even in the absence of external time cues such as the day-night cycle. A number of "clock" genes have been identified as constituting the central mechanism of the brain's timekeeping system, which is central to coordination of metabolic processes, circadian rhythmicity, and sleep regulation (Franken and Dijk 2009; Wulff et al. 2009). To date, researchers have identified, in the pineal gland alone, more than 600 genes whose activities are modulated by the 24-hour sleep-wake rhythm and whose functions influence a diverse range of bodily processes, including inflammation, immunity, transcription, and cell signaling (Bailey et al. 2009).
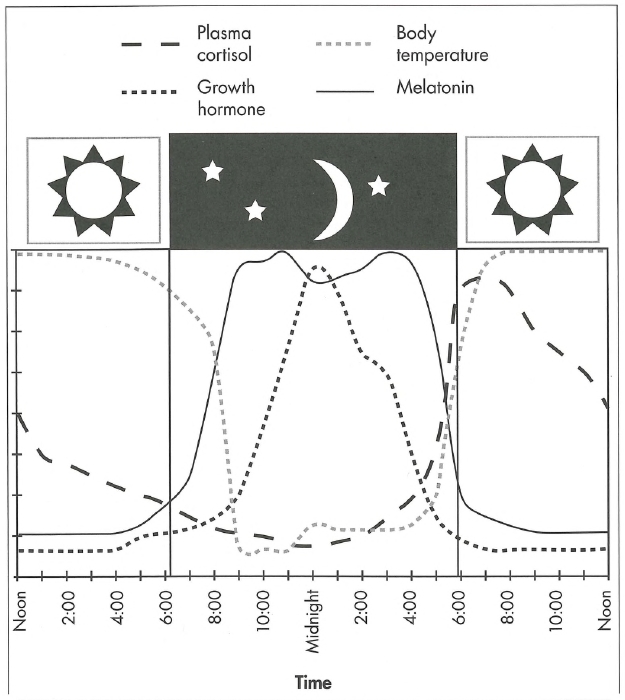
The SCN is the "master clock" that controls circadian timing, and although its basic cycle is a little longer than 24 hours (mean=24.2 hours; range= 23.8-27.1 hours in different individuals), it becomes synchronized to the 24-hour day of humans, primarily through light. Light activates nonvisual retinal photoreceptors, which transmit information via the retinohypothalamic tract serving as the primary influence on the SCN; however, food, temperature, and social influences also impact SCN timing. The SCN transmits timing information to the pineal gland, which produces melatonin. Melatonin production increases in the evening as light decreases, remains elevated during the night, and falls off in the morning. The time at which melatonin increase begins—termed the dim-light melatonin onset (DLMO)—can be a useful biomarker of circadian timing. Maximum sleepiness occurs when body temperature reaches its lowest point (nadir) and melatonin production its highest. Melatonin is only one of a number of hormones with circadian release patterns. Cortisol, the stress hormone, and prolactin, a complex polypeptide with a multiplicity of posttranslational forms and more than 300 biological activities, also demonstrate circadian release patterns (Freeman et al. 2000). Growth hormone peaks at night during deep sleep, but this is a sleep-related rhythm, not a circadian rhythm. Figure 19-1 illustrates the release patterns of these hormones as well as body temperature.
The basic 24-hour rhythm is not present at birth, but rather develops slowly over the first few months of life, first appearing as a greater-than-24-hour free-running rhythm (as new parents learn) and finally becoming entrained to the 24-hour day by about age 16 weeks, the time that most infants can begin sleeping through the night.
About 50% of blind individuals are insensitive to light and thus unable to entrain their circadian system; therefore, they have a free-running period of about 24.2 hours, which causes their sleep-wake cycle to move around the clock such that every several weeks they may sleep through the day rather than the night.
As people age, the influence of light on circadian control diminishes, the period of clock genes may decrease, and melatonin production decreases. These changes may present clinically as sleep-onset insomnia complaints in some elderly individuals, as well as a tendency toward an advanced sleep phase (Singletary and Naidoo 2011). Not all important body rhythms are circadian. Rhythms shorter than 24 hours are termed ultradian and include the REM sleep cycle (approximately 90-120 minutes). Rhythms longer than 24 hours, termed infradian, include, for example, the menstrual cycle (about 28 days).
The various disorders of circadian rhythms are listed in Table 19-1. These circadian disorders most often present as insomnia complaints, although some may present as excessive sleepiness. Sleep-cycle disruption related to jet lag or shift work does not represent a primary disorder of circadian timing but rather a syndrome resulting from behaviorally induced circadian misalignment.
Treatment options for disorders of circadian timing are limited, since there are no effective pharmacological tools yet available. The hypnotic agent ramelteon is a melatonin receptor agonist, but its specific use in circadian rhythm disorders is not yet clear. At this time, proper timing of light exposure and appropriate use of melatonin are the two primary tools available for treatment of disorders of circadian regulation. Exposure to light before the body temperature's nadir will phase-delay the circadian system; exposure to light after the nadir will phase-advance the circadian system. Short-wavelength (blue) light is most effective in circadian timing (Lockley et al. 2006). Melatonin is also used as an adjunct in management of circadian rhythm disruptions, including those related to shift work or jet lag (Cardinali et al. 2006). Many blind individuals can be reentrained with appropriately timed exogenous melatonin (Skene and Arendt 2007).
Several distinct neural systems control wakefulness, NREM sleep, and REM sleep. A basic understanding of these processes and control systems will help the clinician make sense of various sleep complaints that may appear to represent similar problems but in fact result from different mechanisms. When as clinicians we are attempting to treat disorders of sleep and wakefulness, we are often trying to modulate the function of the wake and/or sleep control systems with either upregulation or downregulation. For example, insomnia complaints can often be treated by upregulating sleep control systems (the action of most common hypnotics) or by downregulating wake-promoting systems. It helps to keep in mind what one is trying to do in this regard. Both pharmacological and behavioral strategies are available for these purposes, and these treatments will be discussed in the following sections.
Figure 19-1. Relationship between various hormones and the sleep-wake cycle.
The time scale of the x axis encompasses a 24-hour period from noon to noon, with the sleep period represented by the dark (night) section in the middle. Plasma cortisol secretion begins to increase before morning awakening and peaks in the early morning. Growth hormone secretion (which occurs during Stage III—IV [N3] sleep) peaks early in the night. Melatonin is secreted after dark and is suppressed by light. Body temperature peaks in the late afternoon to early evening and starts to decrease before sleep onset.
Source. Reprinted from Reite M, Weissberg M, Ruddy J: Clinical Manual for the Evaluation and Treatment of Sleep Disorders. Washington, DC, American Psychiatric Publishing, 2009 (Figure 2-10, p. 39). Used with permission.
|
Table 19-1. Overview of circadian rhythm sleep-wake disorders |
||||
| Type | Sleep onset | Treatment | Wakes/alert | Clinical population |
|
Delayed sleep phase |
Later than desired |
Light treatment after lowest core body temperature, melatonin in early evening, hypnotic as needed at bedtime at start of treatment |
Later than desired |
Most common in adolescents and young adults, but may be seen in children |
|
Advanced sleep phase |
Earlier than desired |
Evening light treatment before lowest core body temperature |
Earlier than desired |
Usually the elderly and people with untreated depression |
|
Free running (non-24-hour) |
Moves around the clock |
Melatonin 5-10 mg at desired bedtime |
Moves around the clock |
Blind persons with lack of light exposure due to interrupted retinohypothalamic tract |
|
Irregular sleep phase |
Erratic |
Uncertain—effect of light treatment and melatonin unclear |
Erratic |
Persons with brain diseases, head trauma, or mental retardation |
|
Shift work |
Irregular due to work time |
Modafinil 100-200 mg or caffeine for alertness; melatonin 1-3 mg 1 hour before bedtime; hypnotic (e.g., zolpidem) at bedtime |
Irregular due to work time |
People who work a night shift or rotating shifts |
|
Jet lag (due to circadian misalignment and sleep deprivation) |
Linked to time of origin |
Plot lowest core body temperature at destination and origin; time light and melatonin exposure accordingly Short-acting hypnotic on the flight |
Linked to time of origin |
Persons traveling rapidly across multiple time zones |
Brain areas that support wakefulness include the ascending reticular activating system (ARAS) and cell groups near the mesopontine junction, centered around the pontine and medullary reticular formation that have ascending projections to the forebrain and neocortex and descending projections to brain stem areas that regulate both sleep and wakefulness. The primary neurotransmitters that influence sleep-wake control systems include the catecholamines (dopamine, norepinephrine, and epinephrine) and tryptamines (serotonin and melatonin), histamine (also involved in inflammation), and orexin/hypocretin (also involved in food intake and energy expenditure). Pharmacological agents that modulate these neurotransmitter systems are generally involved in the therapeutic modulation of the sleep-wake system, as discussed below.
Waking is associated with a buildup of adenosine as a result of metabolism; increased adenosine promotes sleepiness and also serves as an activator of the ventrolateral preoptic area (VLPO)-based sleep-promoting systems. The world's most widely used stimulant, caffeine, is an adenosine antagonist.
Wake-promoting systems are extensive and redundant, such that lesions of one system will usually not eliminate the wake-promoting activity of others. During wakefulness, these arousal systems directly inhibit the activity of the sleep-promoting neurons in the VLPO, as discussed later in the section "Neural Systems Promoting Non-REM Sleep." Stress and anxiety result in increased activation or upregulation of wake-promoting systems, as do several drugs of abuse (e.g., amphetamine, cocaine), and a state of hyperarousal results that leads to symptoms of insomnia.
A number of pharmacological agents are used in upregulating wake-promoting systems and promoting arousal. Mono-amine-active agents such as amphetamine and its derivatives, including methylphenidate, are used therapeutically to increase arousal and treat disorders of excessive daytime sleepiness (EDS), as seen with narcolepsy and primary hypersomnia, as well as some medical and psychiatric disorders (e.g., attention-deficit/hyperactivity disorder [ADHD]). Modafinil and armodafinil are newer agents that promote wakefulness through several mechanisms, including increasing the release of two catecholamines, norepinephrine and dopamine; elevating hypothalamic histamine levels; and possibly promoting orexinergic activity.
Downregulation of the ARAS is seen in general anesthesia, and it is possible that some antidepressant agents and atypical antipsychotics sometimes used off label for insomnia may act in this fashion. Low-dose doxepin, a histamine H2 and H2 receptor antagonist, has been approved for insomnia by the U.S. Food and Drug Administration (FDA) and likely acts in this fashion. Most conventional sleep-promoting pharmacological agents (e.g., GABAergic hypnotic agents) discussed in the next sections can also secondarily result in downregulation of the ARAS by virtue of their activation of sleep-promoting systems which in turn decreases ARAS activity.
A number of cognitive-behavioral techniques can be used to downregulate ARAS systems and decrease arousal, thus promoting sleep. These include progressive relaxation, meditation of several types, biofeedback, and components of cognitive-behavioral therapy for insomnia (see Table 19-8, "Sleep restriction and stimulus control," later in chapter). These nonpharmacological approaches to treating insomnia have been shown to be both safe and effective (Morin et al. 2006).
NREM (slow wave) sleep is the state most closely associated with reversal of the neurometabolic effects of wakefulness. NREM sleep is the primary component of Borbely's Process S (the homeostatic sleep drive); the role of REM sleep remains unclear. The primary neurophysiological systems responsible for promoting and maintaining NREM sleep include the output of the predominantly GABAergic and galanergic VLPO and median preoptic (MnPO) nuclei, whose neurons are primarily active during sleep and send inhibitory output to all major cell groups of the hypothalamus and brain stem that participate in arousal (Saper et al. 2005).
Activation of this GABAergic system serves to facilitate and maintain NREM sleep. These sleep-promoting systems are normally inhibited by ARAS wake-promoting activity, but after sufficient sleep debt has accumulated and after the decreasing circadian arousal drive permits, these VLPO systems are activated ("switched on") by the bistable "wake-NREM sleep" switch mechanism (see "Bistable Sleep Switches" below). They then begin to actively inhibit the ARAS systems, permitting entrance into and maintenance of the NREM sleep state. Important to activation of the VLPO-MnPO NREM sleep system is the buildup of adenosine associated with metabolism during the wakeful state. Adenosine appears to be an important homeostatic sleep factor acting through the adenosine A1 and A2 receptors, whose release is triggered by inducible nitric acid synthesis in the basal forebrain secondary to prolonged wakefulness (Sten-berg 2007).
The VLPO is a sexually dimorphic region that is larger and has more cells in males. Men between ages 45 and 60 years show a decrease in cell numbers of about 3% per year until age 60, after which further decreases are not noted. In females, cell numbers decrease until the teen years, then remain fairly stable until after age 50, when a gradual decrease begins; the decrease becomes dramatically more rapid after age 75. Between ages 75 and 85, cell numbers decrease in females at a rate of 4%-8% per year, leading to numbers that are only 10%-15% of the peak seen between ages 2 and 4 (Hofman and Swaab 1989). As implied by these findings, both men and women have increasing difficulty in initiating and maintaining sleep beginning around age 50, but this difficulty stabilizes in men by age 60 while continuing to worsen in women with increasing age. Gaus et al. (2002) have suggested that the shrinkage of the VLPO seen with advancing age may help explain sleep disturbances found in elderly individuals.
The NREM and REM sleep stages and their electroencephalographic and physiological characteristics are shown in Table 19-2. The healthy adult typically transitions through the three NREM sleep stages (N1-N3) before entering REM sleep. Infants, however, often enter REM sleep directly, as do patients with narcolepsy.
Although people are not conscious during sleep, they can respond to external stimuli. For example, neurons in the auditory cortex respond to auditory input during sleep (Issa and Wang 2008), and complex parasomnia behavior, including driving, can occur during sleep, indicating that the brain can respond appropriately to sensory input, as well as regulate motor output, while not conscious of it. Some complex parasomnias (e.g., driving through town asleep) are thought to reflect the release of deep brain central pattern generators normally inhibited during sleep, which permit complex sensory-motor behaviors without cortical awareness (Tassinari et al. 2005, 2009), an example of impaired wake-sleep state control.
|
Table 19-2. Electroencephalographic sleep stages and their physiological correlates |
|||
| Sleep stage | EEG patterns | Physiological correlates | Percentage of total sleep time in adults |
|
N1 |
Loss of waking alpha Increased 5- to 7-Hz theta Occasional high central sharp waves |
Occasional slow rolling eye movement Heart rate and respiration more stable |
5%-7% |
|
N2 |
Sleep spindle (12-14 Hz) bursts K-complexes Higher-voltage mixed theta <20% delta (<4 Hz) |
Heart rate and respiration slower, relatively stable Core body temperature declines EMG decreases |
~50% Most common sleep stage |
|
N3 |
Higher-voltage slowing >75 μV >20% delta Few spindles, rare K-complexes |
Heart rate and respiration slow, stable EMG drops |
~20%-25% Increased in early adolescence Decreased in elderly |
|
REM |
Low-voltage fast activity with mixed theta PGO waves |
Very low EMG with occasional short bursts corresponding to dream activity Rapid eye movements Loss of body temperature control |
~20% 50% in newborns 80% in premature infants Adult levels reached by about age 6 |
Note. EEG=electroencephalographic; EMG=electromyograph; PGO=pontogeniculo-occipital.
The major sleep-promoting system is GABAergic, and most sedative-hypnotic agents approved by the FDA for insomnia treatment are GABAergic agents—that is, either benzodiazepines or the newer omega-1 active nonbenzodiazepine agonists (Table 19-3). The older benzodiazepine hypnotic agents generally nonselectively activate multiple benzodiazepine receptors and thus may have prominent muscle relaxant, anticonvulsant, and anxiolytic properties in addition to their hypnotic actions. They also have in common issues such as tolerance, habituation, and impairment of memory consolidation while the drug is active. Newer nonbenzodiazepine hypnotics are more selectively hypnotic but are not free of other actions.
Another GABAergic agent that promotes sleep is sodium oxybate (sodium γ-hydroxybutyrate), a drug developed under the FDA's orphan drug program as a treatment for narcolepsy.
The most commonly used FDA-approved hypnotic agents are listed in Table 19-3. It is likely that orexin antagonists will be approved in the near future for treatment of insomnia. These agents would of course have an entirely new mechanism of action.
An epoch of NREM sleep is usually followed by a transition to REM sleep. REM sleep is associated with dreaming experiences that would be considered psychotic if they occurred during wakefulness. Abnormalities in REM sleep state control (e.g., narcolepsy) may sometimes be associated with preservation of aspects of the dream state mentation (e.g., visual hallucinations) into wakefulness, which if not recognized may be misinterpreted as psychosis. Some people with narcolepsy are misdiagnosed as having schizophrenia for this reason. In most healthy adults, the time from NREM sleep onset at night to the onset of the first REM period (termed REM latency) is about 90 minutes; however, the latency is shorter in patients with narcolepsy or depression. REM sleep constitutes about 20% of total sleep in adults and occurs in a periodic fashion. REM sleep is accompanied by low-voltage fast electroencephalographic activity, eye movements, loss of skeletal muscle tone (except for muscles of respiration), electroencephalographic ponto-geniculo-occipital waves, and autonomic and body temperature dysregulation. People report dreaming when awakened from this state.
|
Table 19-3. Commonly prescribed hypnotic medications |
|||
| Drug | Mechanism of action | Typical dose | Half-life (hours) |
|
Temazepam |
Bz GABA agonist |
7.5-30 mg |
8-12 |
|
Zaleplon |
Non-Bz GABA agonist |
5-20 mg |
1-1.5 |
|
Zolpidem |
Non-Bz GABA agonist |
2.5-10 mg |
1.5-2.6 |
|
Eszopiclone |
Non-Bz GABA agonist |
1-3 mg |
6 |
|
Ramelteon |
Melatonin agonist |
8 mg |
1-2 |
|
Doxepin |
Antihistamine |
3-6 mg (generic liquid available) |
15 |
|
Sedating antidepressants and antipsychotics as appropriate for coexisting conditions |
|||
Note. Bz = benzodiazepine; GABA=γ-aminobutyric acid.
Transition from NREM to REM sleep is associated with the activation of predominantly cholinergic neuronal systems in the upper pontine regions, with associated decreases in monoaminergic activation. These regions, when activated, produce the physiological accompaniments of REM sleep. Administration of cholinergic agents accentuates REM sleep, whereas drugs promoting monoaminergic activation decrease REM sleep. Alternative REM control mechanisms have been suggested, including activity of GABAergic systems (Fort et al. 2009).
Activation of REM sleep activation is periodic and occurs about every 90 minutes in humans. REM sleep normally appears only on a background of NREM sleep, but REM sleep phenomena may actually continue 24 hours a day in a muted form characterized by varying levels of alertness (Kripke 1972). Most catecholamine agents, as well as many antidepressants, will decrease REM sleep, whereas relatively few agents increase REM sleep, one being reserpine. It is not clear that attempts to therapeutically modulate REM sleep are clinically useful, because deviations from "normal" values in sleep recordings are of uncertain importance.
Transitions from wakefulness to NREM sleep, and vice versa, are controlled by neurophysiological systems acting in a fashion similar to bistable flip-flop switches in electrical engineering (Saper et al. 2010). These systems permit relatively rapid switching to the new state, and then maintain state stability once switched to prevent unstable back-and-forth transitions. The "wake-NREM sleep" switch is influenced primarily by VLPO activity, which when reaching critical levels (facilitated by sleep homeostatic-related adenosine buildup and decrease in circadian arousal drive) begins to actively inhibit arousal systems and triggers a transition into NREM sleep. With loss of VLPO neurons, as occurs in normal aging, the bistable switch mechanism becomes impaired, permitting the switch to "ride" closer to its transition point, resulting in more rapid transitions from wakefulness to sleep, and vice versa, with poor state control, which is often observed in elderly individuals. Orexin/hypocretin neurons contribute materially to the stability of the "wake-NREM sleep" switch, weighting the "wake" side of the switch, whereas loss of orexin neurons (as is seen in narcolepsy) results in state instability with rapid switching between states.
A similar bistable switch mechanism controls transitions from NREM to REM sleep and vice versa; this mechanism consists primarily of pontine neurons at the level of the dorsal raphe. GABAergic neurons within the pons constitute "REM sleep-off" neurons, and neurons in the sublateral dorsal nucleus adjacent to the pons are "REM sleep-on" neurons. These constitute a bistable switch mechanism, with cholinergic activity facilitating the "REM sleep-on" component and monoamine and orexin neurons again contributing to the "REM-off" state. Loss of orexin neurons (as is seen in narcolepsy) contributes to state instability and rapid switching into and out of REM sleep, including "REM sleep-awake state" instability with rapid switching into REM sleep from a wake state (e.g., cataplexy) and sometimes preservation of REM sleep (e.g., dreaming) mentation into wakefulness.
Delta (<4 Hz) sleep is thought to be an index of the homeostatic sleep drive (Process S). In healthy individuals, delta sleep is prominently increased after sleep deprivation. Depression and some medical conditions, including chronic fatigue syndrome and fibromyalgia, are associated with decreases in delta activity, interpreted as a deficiency in homeostatic sleep drive, which may contribute to insomnia-related sleep complaints.
Local delta activity may index synaptic "pruning and tuning" associated with the preceding day's learning; such activity has been found to be regionally increased over areas of cortex experimentally activated during wakefulness prior to sleep onset (Huber et al. 2004). This finding has led to the interesting but not yet well understood concept of "local sleep," wherein (in this case) delta activity is greater over those cortical regions more actively involved in information processing during preceding wakefulness (Huber et al. 2004).
The time from sleep onset at night to completion of the first REM sleep epoch is often termed a sleep period; a normal night's sleep consists of three to five sequential sleep periods. The first sleep periods at night are characterized by a greater amount of delta sleep and relatively little REM sleep, whereas sleep periods in the early morning hours have less delta and longer REM epochs, usually with more intense dream activity. Early sleep periods with their greater delta are more frequently accompanied by NREM-related parasomnias such as sleep terrors and sleepwalking. Later sleep periods with longer and more intense REM epochs are more often characterized by nightmares and REM-related parasomnias. As a result of REM-associated skeletal muscle hypotonia, obstructive apnea events tend to be more prominent during REM sleep.
Slow-wave activity during sleep tends to diminish during the night as the homeostatic sleep drive decreases, whereas REM sleep tends to increase, possibly because it is no longer being suppressed by NREM homeostatic sleep pressure. REM sleep is also homeostatically modulated, and REM suppression will be followed by REM rebound.
Sleep changes dramatically across the life span. The newborn infant, who exhibits a less-well-organized electroencephalogram (EEG), spends approximately 50% of sleep time in REM sleep (premature infants spend even more time in REM sleep—up to 80% at 30 weeks' gestational age). The percentage of time spent in REM sleep approaches adult levels (-20% of total sleep time) during early childhood. Newborns typically have REM sleep at the onset of their sleep periods, shifting to adult NREM-onset sleep periods by about age 4 months. Newborns' sleep is generally about equally divided into active (REM) sleep and quiet sleep, the forerunner of the later-developing Stage N2 and Stage N3 sleep. Stage N2 with sleep spindles and Stage N3 (delta sleep) can usually be identified by about age 3 months.
Total sleep time diminishes with age, ranging from 16 hours per 24 hours at birth to about 9 hours at age 6, about 8 hours at age 12, and typically about 7½ hours in adulthood. Delta activity is very prominent in sleep during early adolescence, and sleepwalking often begins around this time.
Later in adolescence a steep decline occurs in delta sleep that is thought to be associated with age-programmed synaptic pruning. This decline may be an index of brain development or maturation, and Feinberg in 1982 (cited in Feinberg et al. 2006) suggested that a defect in this brain maturational process might underlie some cases of schizophrenia with onset during adolescence. This intriguing hypothesis is still being actively investigated (Boksa 2012). Adolescents also appear to have a physiological circadian phase delay, making it more difficult for them to go to sleep early and arise early in the morning (Carskadon et al. 1993). This phase delay has led some school systems to change their schedules to start classes somewhat later for adolescents.
Typically, delta sleep again begins to decrease with advancing age, possibly due in part to loss of VLPO neurons, decline in aerobic fitness, and perhaps other yet-uncertain mechanisms.
Psychiatrists have long been aware of the close association between sleep and mental illness. Depression is most often accompanied by disturbed sleep. Untreated insomnia can lead to depression. Disturbed sleep may be a harbinger of emerging mental illness. Sleep loss may trigger—or be an early symptom of—a manic episode. Recent research has shown that this bidirectional relationship applies to many other areas of medicine, as well as to overall health. Some examples follow:
2008).
Thus, while we have long known that sleep was essential for life, and that sleep loss was associated with impaired psychological and mental function, we now see emerging the entire complex picture of the intimate relationship between adequate sleep and proper function of multiple specific bodily systems, with significant public health impact. Perhaps it should not be surprising that a state in which we spend about one-third of our existence is so crucial to the effective functioning of the entire organism.
With respect to sleep disorders, the Diagnostic and Statistical Manual of Mental Disorders, 5th Edition (DSM-5; American Psychiatric Association 2013) is one of three major classification systems—the other two being the International Classification of Sleep Disorders, 2nd Edition (ICSD-2; American Academy of Sleep Medicine 2005), which is highly detailed and commonly used by sleep medicine specialists, and the International Classification of Diseases, 9th Edition, Clinical Modification (ICD-9-CM; National Center for Health Statistics 2011), which is used for billing codes in many institutions. Overall, the general classifications are in agreement in these three systems, although specifics vary. A revised third edition of the International Classification of Sleep Disorders is being prepared for release in March 2014, and ICD-10-CM coding will be replacing ICD-9-CM in October 2014.
The conceptualization and organization of sleep-wake disorders have changed substantially in DSM-5 compared with DSM-IV/DSM-IV-TR (American Psychiatric Association 1994, 2000). Previously the sleep disorders were separated by presumed etiology into three nonoverlapping groups: 1) primary sleep disorders (with subcategories dyssomnias and parasomnias), 2) sleep disorders related to another mental disorder, and 3) other sleep disorders (including sleep disorder related to a general medical condition and substance-induced sleep disorder). In DSM-5 the individual disorders are separated on the basis of comorbidity and coexisting conditions rather than on any presumptions regarding causation.
DSM-5 sleep-wake disorders now constitute 10 separate disorders or disorder groups: insomnia disorder; hypersomnolence disorder; narcolepsy; breathing-related sleep disorders; circadian rhythm sleep-wake disorders; parasomnias including NREM sleep arousal disorders, nightmare disorder, REM sleep behavior disorder, and restless legs syndrome; and substance/medication-induced sleep disorder. In the insomnia domain, criteria for an insomnia diagnosis have been changed (with primary insomnia replaced by insomnia disorder) and subtypes of circadian rhythm disorders expanded, with jet lag removed. In the hypersomnia domain, narcolepsy has been separated from the hypersomnia group, as its association with hypocretin deficiency has been identified. The breathing disorder group has been more clearly specified and includes obstructive sleep apnea hypopnea, central sleep apnea, and sleep-related hypoventilation. The central apnea and hypoventilation groups have several subtypes for clarification.
Two new diagnoses have been added, REM sleep behavior disorder and restless legs syndrome, to limit use of the "unspecified" classification (including the former "not otherwise specified [NOS]" diagnoses in DSM-IV).
In addition to diagnostic criteria, DSM-5 provides details on differential diagnosis, as well as available data on prevalence, development and course, risk and prognostic factors, functional consequences, comorbidity, diagnostic markers (when available), and other information relevant to understanding the disorders. Associated codes for ICD-9-CM and ICD-10-CM are provided, and relationships with International Classification of Sleep Disorders diagnostic categories are discussed.
Once a diagnosis has been made, where does the clinician go to find treatment options? This chapter does not attempt to deal with the specifics of treatment for all of the various sleep disorders, nor does DSM-5. Extensive discussion of treatment options can be found in major textbooks of sleep, such as Principles and Practice of Sleep Medicine, 5th Edition (Kryger et al. 2011), and a recent textbook devoted to treatment (Barkoukis et al. 2012). Shorter reviews of treatment options can be found in volumes such as Clinical Manual for Evaluation and Treatment of Sleep Disorders (Reite 2009). One good treatment resource is the publications prepared by the American Academy of Sleep Medicine (www.aasm-net.org). These Practice Parameters, Clinical Guidelines, and Best Practice Guides cover the general areas of insomnia, hypersomnia, circadian rhythm disorders, parasomnias, sleep-related breathing disorders, sleep-related movement disorders, and pediatric sleep disorders. Efforts are made to update these publications on a periodic basis.
In this section we present an approach that we believe will help non-sleep clinicians to identify and either treat or appropriately refer common sleep disorders encountered in their practices.
Sleep disorders derail health and shorten longevity. They are highly prevalent, affecting an estimated 50-70 million individuals in the United States alone. In the general population, insomnia symptoms are present in up to 33% of individuals, obstructive sleep apnea in about 5% (although 26%-32% have symptoms suggesting risk for OSA), and restless legs syndrome in 5%-15% (Senthilvel et al. 2011).
Although widespread, sleep disorders often remain unexplored if not asked about by clinicians or brought up by patients (Roth et al. 2010; Wells and Vaughn 2012). Symptoms can be expressed directly by patients ("I can't sleep") or complained of by others ("her snoring/his thrashing keeps me up"); at times, the possibility of a sleep problem can be inferred from the presence of other diagnoses known to coexist with disordered sleep, such as depression, hypertension, stroke, or other cardiac disease. However, even when a sleep disturbance is known to be present, many clinicians lack a framework for conducting a thorough evaluation.
The primary symptoms of disordered sleep—insomnia, excessive daytime sleepiness, and disturbed or disturbing sleep behavior—are usually not pathognomonic for a specific diagnosis and must be further parsed and explored (Slater and Steier 2012). For instance, some people who claim to be sleepy may actually be fatigued—unable to either sleep at night or nap during the day. Other people are "night owls" (i.e., have a delayed sleep phase circadian rhythm disorder) who do not sleep in tune with their circadian arousal systems; these individuals can develop EDS, insomnia, or both (Bootzin and Epstein 2011; Ebben and Spielman 2009; Reid and Zee 2009).
The bidirectional relationship between sleep disorders and psychiatric illness—coupled with the fact that sleep disorders often coexist with other medical, psychiatric, and sleep problems—creates a host of diagnostic pitfalls. For example, problems can arise when a clinician assumes that one disorder (e.g., comorbid depression) "causes" another (e.g., insomnia). Although depression can indeed cause sleepiness and insomnia, depression and insomnia may coexist independently and be mutually influencing; therefore, insomnia and depression each require a separate assessment (Bootzin and Epstein 2011). An insomnia disorder may have multiple roots; for instance, it may be precipitated by OSA or the circadian misalignment of sleep phase delay. In fact, it is not unusual for a patient to have all three of these conditions—OSA, insomnia disorder, and a circadian rhythm disorder-delayed sleep phase type.
Problems also arise when a clinician, having identified "the cause" of a patient's insomnia, assumes that further diagnostic evaluation is not necessary. This is an error, since more than one condition is frequently present (Ebben and Spielman 2009; Pigeon et al. 2012; Slater and Steier 2012; Soreca et al. 2012). As John B. Hickam stated (in the medical profession's counterargument to Occam's razor that has become known as Hickam's dictum), "Patients can have as many diseases as they damn well please."
Approximately 80 sleep-wake disorders have been identified at present (American Academy of Sleep Medicine 2005). The DSM-5 sleep-wake classification encompasses considerably fewer disorders, from which we have chosen seven to demonstrate our clinical approach:
We strongly recommend that the first four disorders listed—delayed sleep phase circadian rhythm disorder ("night owl" syndrome), OS A, insomnia disorder, and RLS—be considered in all patients who present with sleep complaints, regardless of what other medical or psychiatric diagnoses are present. When symptoms suggest, the last three listed disorders should also be explored. Of high clinical importance is the fact that four of these seven disorders—OS A, RLS, sleepwalking, and REM sleep behavior disorder—can be precipitated or worsened by commonly prescribed medications.
The DSM-5 diagnostic criteria for each of these seven disorder categories are included in the Appendix to this chapter.
|
The Four-Step Evaluation |
|
Although all patients should be screened for sleep disorders, such screening is particularly important if medical or psychiatric disorders known to coexist with sleep disorders are present. Such disorders include ADHD, cardiovascular disease, hypertension, diabetes, and depression. The following screening questions can be asked to determine the need for further exploration:
Augmenting the history by interviewing someone familiar with the patient usually provides valuable insights. Reports of snorts or gasps in a patient who denies snoring should prompt serious consideration of breathing-related sleep disorders, whereas unusual motor behaviors raise the possibility of an NREM sleep arousal disorder (e.g., sleepwalking), a REM sleep behavior disorder, GSA, or nocturnal seizures.
"Sleepiness" is a common complaint of patients who cannot stay awake but also of those who cannot sleep when given the chance. The former is true sleepiness (EDS); the latter is fatigue due to physiological hyperarousal, which is often accompanied by lack of energy, motivation, and mental clarity. The differential diagnosis for true sleepiness is not the same as that for fatigue (Table 19-4).
People's perceptions of sleepiness can be unreliable; some people may tolerate high degrees of sleepiness without actually "knowing" that they are sleepy. In such situations, behavioral symptoms can help identify sleepiness (Table 19-5). In contrast, electrophysiological recordings are quite sensitive and can reliably detect sleepiness. Another useful tool in this regard is the Epworth Sleepiness Scale (Johns 1991; Table 19-6), a self-administered questionnaire in which the respondent is asked to rate, on a 4-point scale (0-3), his or her chances of dozing off in each of eight different situations. The summed score provides a measure of impairment and is helpful in distinguishing fatigue from true sleepiness (Beaudreau et al. 2012; Sil and Barr 2012). Some clinicians use an Epworth score of 8 as the upper limit of "normal"; others consider a score greater than 10 as an indication of pathological sleepiness. High Epworth scores may indicate EDS, possibly resulting from a breathing-related sleep disorder (Rosenthal and Dolan 2008), insufficient sleep opportunity, or a circadian rhythm sleep-wake disorder. However, a low score in the "normal range" (e.g., 2-6) can be misleading, because many patients with OSA, for instance, do not feel particularly sleepy. Epworth scores are only one piece of the diagnostic puzzle.
Very low Epworth scores (typically 0-2) may indicate that the patient is unable to relax day or night, perhaps as a result of hyperarousal secondary to anxiety, depression, or "sleep worry" (the latter is the basis of learned or conditioned psychophysiological insomnia—i.e., insomnia disorder). Hyperaroused individuals have higher core body temperatures, faster EEGs, and elevated cortisol levels. They have trouble disengaging from the world as they try to sleep. Hypnotic medications often fail to work in such patients (Bootzin and Epstein 2011).
|
Table 19-4. Causes of excessive daytime sleepiness (EDS) and fatigue |
||
|
Sleepiness (behavioral symptoms of sleepiness3; elevated score on Epworth Sleepiness Scaleb) Obstructive or central sleep apnea: 70% of patients with obstructive sleep apnea are sleepy, whereas 30% are not; central sleep apnea may be precipitated by opioid use Circadian rhythm disorders (e.g., delayed sleep phase, shift work, jet lag) Insufficient sleep opportunity Narcolepsy Idiopathic hypersomnia Head injury Depression: especially seasonal, atypical, and bipolar (but other causes should be considered) Drug use or withdrawal Restless legs syndrome Medical illness (e.g., renal or hepatic failure, brain tumors, neurodegenerative disorders) Fatigue (low score on Epworth Sleepiness Scaleb) Anxiety and depression Conditioned/learned insomnia |
||
aSee Table 19-5 for behavioral symptoms of sleepiness.
bSee Table 19-6 for Epworth Sleepiness Scale questions.
|
Table 19-5. Behavioral symptoms of sleepiness |
||
|
||
The Epworth Sleepiness Scale may yield false negatives but rarely false positive scores. About 30% of patients with OSA, a common cause of EDS, have normal Epworth scores. Some patients in this group may have developed conditioned insomnia and are experiencing hyperarousal from sleep disruption secondary to their OSA. True EDS can also be present in patients experiencing winter depression, atypical depression, or the depressed phase of bipolar illness. The bottom line: if the Epworth score is elevated, the clinician must determine why. Epworth Sleepiness Scale scores should be evaluated carefully within the patient's clinical context, including behavioral indications of sleepiness or fatigue.
Although not curative in itself, establishing healthy sleep habits is generally part of sleep disorder treatment (Schutte-Rodin et al. 2008). Factors that may contribute to or cause EDS or insomnia should be identified and remedied where possible. For example, does the patient
|
Table 19-6. Epworth Sleepiness Scale |
||
|
How likely are you to doze off or fall asleep in the following situations, in contrast to just feeling tired? This refers to your usual way of life in recent times. Even if you haven't done some of these things recently, try to work out how they would have affected you. Use the following scale to choose the most appropriate number for each situation: 0 = would never doze 1 = slight chance of dozing 2 - moderate chance of dozing 3 = high chance of dozing Sitting and reading ____ Watching TV ____ Sitting, inactive in a public place (e.g., a theater or a meeting) ____ As a passenger in a car for an hour without a break ____ Lying down to rest in the afternoon when circumstances permit ____ Sitting and talking to someone ____ Sitting quietly after a lunch without alcohol ____ In a car, while stopped for a few minutes in traffic ____ TOTAL ____ SCORING: ____ |
||
Availability and Conditions of Use. The developer and copyright holder of the Epworth Sleepiness Scale, Dr. Murray Johns, permits use of the scale by individual people (including clinicians and researchers) free of charge. The Epworth Sleepiness Scale is downloadable from the Web Source shown below, from which complete administration/scoring instructions and methodological data are also available.
Source Reference. Johns MW: "A New Method for Measuring Daytime Sleepiness: The Epworth Sleepiness Scale." Sleep 14:540-545, 1991.
Web Source. Official Web site of the Epworth Sleepiness Scale by Dr. Murray Johns (http://epworth-sleepinessscale.com/1997-version-ess).
A widely held misconception is that "sleep hygiene" is the treatment for sleep complaints, not just an important part of a comprehensive approach. A thorough evaluation requires assessment of which disorders are actually present.
Core body temperature (CBT) is tightly linked with, and a proxy for, the circadian timing system that controls our sleep-wake cycle (endogenous rhythm ~24.2 hours) and oscillates independently of whether we are asleep or awake. If awake for long periods, we feel more or less alert due to these oscillations. We sleep best as the temperature drops (here around midnight) and awake approximately 2 hours after lowest CBT begins to rise. We sleep poorly when not in tune with these rhythms. Night owls' rhythms are delayed and occur later in the 24-hour day-night cycle. Their CBT may begin to drop at 2 A.M. or 3 A.M. and rise in the late morning or afternoon. If night owls sleep in tune with their rhythms, they are out of tune with social demands. If not, they develop insomnia, excessive daytime sleepiness, or both. Shift workers and those with jet lag also develop sleep problems when trying to sleep or stay awake at the wrong times for their internal rhythms.
Circadian alerting rhythms wax and wane on their own schedules, essentially unaffected by our efforts to sleep or stay awake. When people do not follow these rhythms, symptoms of insomnia or EDS arise and may persist for years before misalignment of the sleep schedule with the circadian timing system is identified as their cause (Reid and Zee 2009; Schaefer et al. 2012; Schwartz 2010).
Sleep is efficiently generated when our sleep debt, which increases in a linear fashion while we are awake, intersects with our circadian arousal system as it starts to "quiet" at night. We sleep best when our CBT drops (and our arousal system quiets), and we wake spontaneously about 2 hours after our CBT begins to rise. Insomnia develops if we try to initiate sleep before our CBT begins to drop, while our arousal system is still set to "loud"; EDS develops when our sleep period is truncated by the requirement to wake at a specific time, and that hour arrives before we have gotten enough sleep.
Misalignment of alerting rhythms with social demands is easy to identify in people who do shift work or have jet lag; it is harder to spot in people who are early risers (so-called larks) or who stay up late (night owls). Rhythms are advanced to earlier times in about 1% of the population (larks), usually with onset in middle age; rhythms are delayed in up to 15% of adolescents and in somewhat fewer adults. When people with advanced rhythms are able to sleep in tune with their circadian systems, they fall asleep early, wake early, and sleep normally. However, if they stay awake to meet social demands, they still wake early and may develop EDS due to sleep lack, and their "early morning awakening" might be misattributed to depression.
Night owls, in contrast, sleep normally when they are in tune with their delayed clock but can develop "insomnia" if they attempt to sleep at earlier, socially appropriate times) and/or EDS if they awaken before their alerting rhythms begin to rise and adequate sleep has been obtained. Circadian rhythm sleep-wake disorder—delayed sleep phase type is one cause of "bedtime difficulties" in children; however, such children are not really being "difficult"—they are just unable to fall asleep. These children put to bed before biologically ready for sleep also may lie in bed for hours, their minds active, developing fears of the dark and be diagnosed with anxiety when the issue is precipitated by parental behavior.
Although patients with mood disorders such as major depression, bipolar disorder, and seasonal depression seem to have a higher incidence of sleep phase delay, mood issues are not always present in night owls. Of course, "ersatz" sleep phase delay may be due to purely behavioral issues as well; when social demands arise, individuals with behaviorally induced sleep phase delay have little difficulty adjusting their sleep schedules to more appropriate times.
Alerting rhythms are under the control of clock genes and light exposure (Roenneberg et al. 2007). For individuals who are in constant darkness, these rhythms typically wax and wane in 24.2-hour cycles but are reset to the 24-hour photoperiod by morning light exposure after the lowest CBT, which occurs about 2 hours before spontaneous wake time. Individuals sleep best by initiating sleep when their sleep debt is high and their alerting signals are waning. People wake spontaneously when sleep debt—lowered during sleep—intersects with the rising alerting system, about 2 hours after their lowest CBT. It is difficult to sleep on a rising CBT, a problem experienced by night shift workers who try to sleep during the day. It is almost impossible to initiate sleep during the few hours before CBT begins to fall, when alerting systems are at their "loudest." This difficulty is experienced by night owls who try to initiate sleep at more conventional bedtimes, despite the fact that their CBT will not begin to drop until hours later (Reid and Zee 2009).
Morningness-eveningness questionnaires are available (Morgenthaler et al. 2007) but are cumbersome for clinical settings. Because measurement of circadian markers (i.e., dim-light melatonin onset and offset) is not yet clinically available, questioning about circadian propensities is necessary. A series of probes typically suffices:
It is sometimes easier to identify circadian wake times (the time of day at which a patient becomes fully alert) and work back by 7-9 hours to get an idea of likely circadian sleep-onset times. Confusion arises when patients have high sleep debts, which can occasionally induce "normal" sleep-onset times. Some patients report gradually drifting to later sleep-onset times and ultimately not sleeping at all. The latter situation could be due to a manic episode or hypothalamic malfunction but likely occurs because of light exposure before the lowest CBT, which shifts the clock later. Other factors might be greater sensitivity of the phase-delay portion of the phase-response curve or lack of exposure to "morning light," which would advance rhythms to an earlier time. Some patients get no daylight at all; their clock never is reset to 24 hours and appears to "run free" on a 24.2-hour schedule. True free-running sleep disorder might be present but is rare.
Successful treatment of circadian misalignment depends on properly timed exposure to light and a "physiological" dose of melatonin. Effective treatment of circadian rhythm disorders can be provided by clinicians other than sleep specialists (Morgenthaler et al. 2007). Melatonin taken before lowest CBT and light exposure obtained after lowest CBT resets the alerting system to an earlier time; light exposure before lowest CBT and melatonin after pushes the alerting system to a later schedule. If the retinohypothalamic tract (the photic pathway involved in the circadian rhythms of mammals) is intact, blind people can be treated with light. Patients in whom this tract is interrupted and "free running" (i.e., controlled by the 24.2-hour endogenous rhythm) can be reentrained with melatonin given at bedtime.
The recommended treatment regimen for the delayed sleep phase type of circadian rhythm sleep-wake disorder is as follows:
Finally, recommend that the patient keep a log detailing 1) timing of sleep, 2) light exposure, and 3) melatonin ingestion. Light exposure at "alarm clock" wake time, before lowest CBT, delays rhythms and is one cause of treatment "failures." We evolved to sleep best in tune with our circadian rhythms. Deviation from an "in tune" schedule is a common cause of poor or insufficient sleep and of sleepiness when we need to be awake.
OS A is always part of the differential diagnosis of insomnia and EDS and should be considered when conditions known to be associated with OSA are present. OSA is present in 2% of children, is common in genetic disorders such as Down syndrome, and is present in up to 15% of middle-aged adults and more than 20% of elderly persons. OSA is associated with problems in addition to insomnia and EDS—including depression and treatment-resistant depression; attentional problems in children (sometimes diagnosed as ADHD); impaired cognition; declines in work and school performance; increased risk of cardiovascular heart disease, hypertension, and stroke; reflux and heartburn; morning headaches; nocturia; erectile dysfunction; and decreased libido—and is often present in sleepwalkers and people with diabetes (Kasai et al. 2012; Slater and Steier 2012; Soreca et al. 2012; Viera 2012).
Key symptoms of snoring and daytime sleepiness are often not obvious because many patients with OSA are not subjectively sleepy and some hardly snore. For instance, women may present with insomnia and fatigue rather than loud snoring or sleepiness. Symptoms may be subtle in children, sometimes manifesting as behavioral, attentional (ADHD), or sleep problems. Other symptoms that should trigger suspicion of OSA in children include mouth breathing, poor speech articulation and swallowing, failure to thrive, developmental delays, and return of enuresis after being dry.
Obesity is the strongest risk factor for OSA, and the prevalence of OSA is much higher in patients with cardiac or metabolic disorders. Psychotropic medication-induced weight gain may also contribute to the development of OSA. Hypothyroidism, Cushing's syndrome, acromegaly, cerebral palsy, Down syndrome, Prader-Willi syndrome, neuromuscular disease, Parkinson's disease, asthma, and sicklecell disease also increase the risk.
Morbidity and mortality from OSA increase with age, peaking in the mid-50s. Despite the disorder's connection with obesity, not all people with OSA are overweight; 30% are thin and fit. Facial structure (narrow chin, heart-shaped face, and overbite), genetics, and family history play a role (the incidence is twice as high in first-degree relatives of those with the syndrome).
Although risk increases with obesity and male gender, as well as with a neck circumference >16 inches in women and >17 inches in men, adults and children of all shapes and sizes may have symptoms for many years yet remain undiagnosed until they come to clinical attention because of one of the downstream effects of sleep apnea (e.g., heart disease, hypertension, behavioral problems) or because of fatigue or insomnia, weight gain, or "spousal arousal" (i.e., disruption of a bed partner's sleep) (Brostrom et al. 2012; Erichsen et al. 2012; Slater and Steier 2012).
OSA is characterized by repetitive episodes of partial to complete obstruction of the upper airway during sleep. The disorder is misnamed, because complete airway obstruction is not necessary for pathological changes to occur. A minor collapse increases the work of breathing and causes a shift of brain waves to faster rhythms, termed a respiratory effort-related arousal; a somewhat greater collapse results in hypopneas, which are associated with a drop in oxygen or an arousal; finally, a complete collapse is an apnea. People prone to OSA have airways that collapse with less effort, possibly as a result of physiological characteristics (e.g., narrow airway, thick neck, large tongue, small or receding chin, larger fat pads in the neck, large tonsils and adenoids) or because of central control issues.
Increased work of breathing exists on a continuum from primary snoring (no evidence of collapse) to total collapse. Hypoxic stress precipitates a proinflammatory response with oxidative stress and endothelial dysfunction, which can lead to cardiovascular disease, stroke, sudden cardiac death, hypertension, and pulmonary hypertension. Primary snoring is not necessarily benign. Some children diagnosed with primary snoring were found to have abnormal nighttime blood pressure (Li et al. 2009; Weber et al. 2012). There is evidence that mothers who snore during pregnancy are at greater risk of having babies with lower Apgar scores and birth weights (Ibrahim and Foldvary-Schaefer 2012).
Asking about snoring should be part of all medical and psychiatric evaluations, as should consideration of facial structure (e.g., heart-shaped face, receding chin, crowded airway, overbite). If OSA is suspected, a polysomnogram currently is the only definitive way to assess its presence; nocturnal oximetry and questionnaires can result in false negatives. If the diagnosis is uncertain, the clinician should refer the patient to a sleep laboratory that scores respiratory effort-related arousals, a measure that increases diagnostic sensitivity.
If sleep-related breathing disorder is suspected, the workup and treatment might best be initiated and managed by a physician trained in sleep medicine. In children, removal of tonsils and/or tissues obstructing the airway is often necessary (Marcus et al. 2012a, 2012b); in adults, continuous positive airway pressure (CPAP) is usually the preferred method of treatment (Fleetham et al. 2011). Success during the early weeks of CPAP treatment is critical for long-term adherence. Patients need frequent contact with someone expert in mask fitting, pressure adjustment, and smart card downloads to track adherence and clinical response. Use of hypnotics for the first few weeks of CPAP therapy is sometimes helpful. Some patients with mild to moderate apnea can be managed with custom-made oral appliances that help keep the airway open. The fitting of these devices is the province of sleep dentistry and requires a follow-up sleep study. Treating the breathing disorder does not necessarily mean that the sleep problem will resolve. Patients may have developed other disorders, such as insomnia, that will need independent assessment and management.
Insomnia is the most complex sleep disorder that the non-sleep clinician will encounter. It is the most frequent sleep complaint; it becomes insomnia disorder when patients experience daytime symptoms, thereby turning it into a 24-hour-a-day problem. The timing of sleeplessness may shift over time, with problems of sleep initiation, continuity, and early-morning awakening present in the same patient at different points. Patients who experience true daytime sleepiness (as indicated by an elevated Epworth Sleepiness Scale score) are likely different from those who are fatigued and unable to sleep day or night (except, sometimes, when away from home). It is useful to think of such patients as having a type of "anxiety disorder" about sleep. Insomnia of more than 1 year's duration increases the risk of new-onset major depression; the presence of insomnia and nightmares increases suicide risk.
Insomnia usually coexists with other conditions: psychiatric, sleep, and medical. Although comorbid conditions may be mutually influencing (Table 19-7), each requires individual diagnostic attention even if is assumed that one is causing the other (Morin and Benca 2012; Schutte-Rodin et al. 2008). Patients may report experiencing fatigue, sleepiness, cognitive or mood disturbances, and reduced social, academic, and work effectiveness.
The most common form of insomnia is transient, lasting from a day or two up to several weeks, with known causes including stress of all types, excitement, ascension to high altitudes, and circadian misalignment (e.g., due to jet lag or shift work). Such problems rarely come to the attention of clinicians in early stages or may respond to a brief course of a hypnotic (see Table 19-3, "Commonly prescribed hypnotic medications"). Transient insomnias are also common during the late luteal phase of menstruation, in women with early menses, and in perimenopausal and postmenopausal women.
Of greater concern are chronic insomnias that last for weeks, months, or years, which can be associated with medical (e.g., cardiovascular disease, hypertension, type 2 diabetes) and psychiatric (anxiety, depression, elevated suicide risk, substance use) morbidity (Pigeon et al. 2012). All patients with insomnia, regardless of the initial cause, are at risk of developing an element of conditioned (psychophysiological) insomnia, which involves physiological hyperarousal due to negative conditioning and intense worry about lack of sleep.
Patients with chronic insomnia enter sleep faster, sleep better and longer than they think, and may even be asleep when they feel they are awake—a condition called sleep state misperception. One of the first observations of this phenomenon by Perlis et al. (1997) noted that these patients' EEG activity is faster than normal at sleep onset, which blurs the distinction between sleep and wakefulness. Patients secrete high levels of stress hormones, and their brain metabolism is abnormally high, as is their heart rate and sympathetic nervous system activity during sleep. Chronic day and night hyperarousal likely plays a role in patients' increased risk of developing medical disorders such as depression, hypertension, or cardiac disease (Bonnet and Arand 2010). Hyperaroused patients commonly experience multiple failed medication trials but do respond to mindfulness-based interventions and cognitive-behavioral therapy for insomnia (including sleep restriction and stimulus control).
|
Table 19-7. Disorders that may coexist with insomnia |
|
Sleep disorders Circadian misalignment Obstructive/central sleep apnea Narcolepsy Restless legs syndrome Medical disorders Gastroesophageal reflux disease, nocturia, pain Movement disorders Fibromyalgia, chronic fatigue syndrome: disrupt sleep/circadian systems Dementia: decreases sleep-wake control Psychiatric disorders Anxiety: increases hyperarousal Posttraumatic stress disorder: causes insomnia and parasomnias Depression Atypical depression: can cause hypersomnia Bipolar disorders: can cause impaired circadian control Substance misuse: can cause insomnia, sleep disruption |
Figure 19-2 presents a "road map" to guide evaluation and treatment of insomnia. Identification of Spielman's "3 Ps"—the predisposing, precipitating, and perpetuating factors for insomnia—provides the framework for generating a list of potential causes, identifying coexisting conditions, and planning or referring for treatment. One or all of the Ps may require clinical attention (Ebben and Spiel-man 2009).
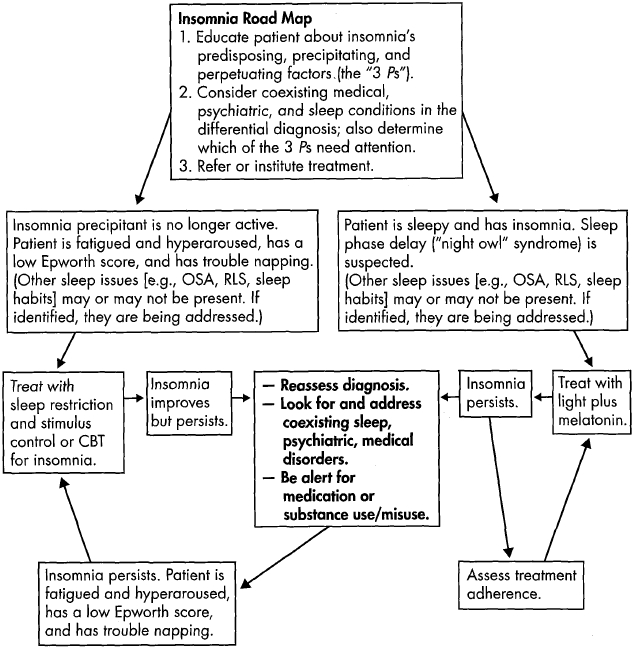
Figure 19-2. Insomnia road map: a guide to evaluation and treatment.
Note. More than one problem may impact sleep; for example, the patient may be a night owl with obstructive sleep apnea who develops hyperaroused/psychophysiological insomnia and struggles with bipolar depression. When identified, all problems are amenable to treatment. CBT=cognitive-behavioral therapy; OSA=obstructive sleep apnea; RLS=restless legs syndrome.
Assuming that coexisting disorders have been or will be addressed, the general approach to patients with insomnia is as follows:
|
Table 19-8. Sleep restriction and stimulus control |
||
|
||
RLS is rarely mentioned by patients, but its prevalence is 3%-15% and appears to be increasing, possibly because advertisements for dopamine agonists have raised public awareness of the symptoms, enabling sufferers to identify the cause of their nighttime discomfort. Depending on intensity and frequency, RLS may adversely affect sleep and have serious health consequences. RLS can be primary and familial or secondary due to variety of other factors, including medications. It is one of several potentially iatrogenic sleep disorders, a category including NREM sleep arousal disorders (e.g., sleepwalking), REM sleep behavior disorder, and OS A, all of which can be precipitated or worsened by psychotropic medications (Salas et al. 2010; Sieminski 2012).
RLS is a waking diagnosis and needs to be differentiated from nervous foot shaking (unless this relieves leg discomfort) and periodic leg movements in sleep (PLMS) (although PLMS is found in the majority of patients with RLS). RLS is a neurological disorder characterized by the urge to move parts of one's body—most often the legs and/or arms—to relieve uncomfortable or unusual sensations. Patients have a hard time describing their symptoms, which exist on a spectrum of mild to severe and which occur when patients are awake in restful situations (e.g., reading, studying, trying to sleep) and usually at night (Innes et al. 2011). People with RLS may use phrases such as "runny legs" or "crawly worms" when describing symptoms; 20% of patients report pain. When seen in children, RLS may be misdiagnosed as "growing pains," and clinicians may overlook the role of RLS in precipitating attentional problems (Owens 2011). Seventy to eighty percent of patients with RLS demonstrate PLMS, whereas 30% of patients with PLMS report symptoms of RLS. PLMS is observed on nocturnal sleep studies or noted by bed partners and is thought to correlate poorly with sleep disturbance or daytime symptoms. RLS also needs to be differentiated from akathisias attributable to antipsychotics or dopamine antagonists. RLS can increase the sleep pressure and sleep disruption that play a role in NREM sleep arousal disorders—sleep terrors, sleepwalking, and confusional arousals in the genetically predisposed.
Approximately 60% of RLS cases are thought to be familial, transmitted as an autosomal dominant trait with variable penetrance. Dopamine abnormalities likely play a role, and RLS is associated in some patients with low levels of serum ferritin, which is a cofactor in dopamine metabolism. Risk of secondary RLS in creases with age and is higher during pregnancy (third trimester). Risk is also increased in women; in persons with iron deficiency anemia; in vegetarians; in persons with renal disease, neuropathies, ADHD, or Parkinson's disease; and in those who abuse alcohol, use excessive caffeine, or smoke (Ibrahim and Fold-vary-Schaefer 2012). Medications that precipitate or worsen RLS include most psychotropics—selective serotonin reuptake inhibitors (SSRIs), tricyclic antidepressants (TCAs), antipsychotics, and lithium—and also antihistamines. Bupropion does not cause RLS or PLMS, likely because it increases dopamine.
Treatment of RLS is not necessary if symptoms rarely impact sleep. If symptoms warrant, the clinician should identify and attempt to remove offending agents such as nicotine, alcohol, or psychotropic medications.
For all patients with RLS, the clinician should obtain a serum ferritin level, which provides a measure of body iron stores. Iron needs to be replaced if levels are 50 ng/mL or less (although a level of 50 ng/Ml will be reported as "normal" by laboratories). Some experts replace iron at levels well above that level, since cerebrospinal fluid ferritin levels can be lower than peripheral values. The causes of low levels need to be identified (e.g., diet). Typically, ferrous sulfate 325 mg with 100 mg vitamin C (which promotes absorption) is taken twice a day if tolerated. It should not be taken with meals. Ferritin levels should be retested every 3-4 months to avoid overtreatment.
Nonmedical treatments for RLS include massage, warm baths, relaxation, exercise, and reduction of caffeine and alcohol intake. Mindfulness-based meditation has helped some patients.
Medication classes that have been found useful for treating RLS include opiates (e.g., methadone), anticonvulsants (e.g., gabapentin), benzodiazepines (e.g., clonazepam), and dopamine agonists (e.g., ropinirole). Although still widely prescribed, dopamine agonists can cause symptom rebound and augmentation (Salas et al. 2010); for this reason, some sleep clinicians prefer not to prescribe dopamine agonists.
Of the three disorder categories reviewed in this step, two—NREM sleep arousal disorders and REM sleep behavior disorder—are parasomnias (abnormal movements and behaviors associated with sleep). These disorders are potentially dangerous, can be precipitated by psychotropic medications, and may be confused with one another (Kotagal 2009; Mahowald and Schenk 2004; Mahowald et al. 2011; Postuma et al. 2012).
NREM sleep arousal disorders affect about 17% of children and typically resolve in early adolescence; these disorders are also present in about 4% of adults. In DSM-5, NREM sleep arousal events are separated into two types, which may blend into each other: sleepwalking and sleep terrors (confusional arousals are still considered by some to be part of this spectrum). Episodes typically arise out of slow-wave sleep during the first third of the sleep period, when NREM sleep predominates. Patients are hard to wake, although their eyes are open; in the morning, they have little or no recall of their activities.
Sleep-related eating (SRE) and sleep-related sexual behavior (sexsomnia) are likely subtypes of sleepwalking. These disorders may be activated by sedative-hypnotics and respond to dose reduction. SRE should not be confused with night eating syndrome (NES), an ill-defined condition in which awake patients binge-eat late at night. It is thought that this behavior may be due in part to circadian delays; treatment may involve sertraline and raising nighttime melatonin secretion.
NREM sleep arousal disorder symptoms exist on a continuum: confusional arousals (sitting up, eyes open, talking), sleep terrors (usually beginning with a bloodcurdling scream), and sleepwalking all can occur in the same patient. During episodes, frontal lobes are "offline"; patients appear to be awake, with open eyes, and may be able to converse, but awareness is not present. Sleepwalkers have climbed or fallen out of windows, jumped off roofs, driven cars, and attacked those who got in their way or tried to wake them. In the morning, sleepwalkers are partially or totally amnestic for nighttime events.
The pathophysiology of NREM arousal disorders is poorly understood. Fragmented slow-wave sleep, sleep state dissociation, and genetic factors are part of the story (Zadra et al. 2013). A family history is found in about 80% of patients. Arousal events occur during the first third of sleep, when slow-wave sleep (N3, which can be abnormally fragmented) predominates. They can also arise out of N2 sleep, especially if patients are extremely sleep deprived (Provini et al. 2011). In addition, the concept of "local sleep"—that sleep and wakefulness are not mutually exclusive but can coexist, a phenomenon that is seen in dolphins, for instance—is increasingly accepted (Huber et al. 2004).
Pressman's (2007) predisposing, priming, and precipitating factors can be a useful tool in evaluating NREM sleep arousal episodes. Genetically predisposed individuals are primed to sleepwalk as a result of conditions such as increased sleep pressure (from sleep deprivation, schedule changes, or poor sleep hygiene) or substances such as alcohol or medications (e.g., propranolol, antiarrhythmics, sedatives, hypnotics, antidepressants, lithium, L-dopa, antipsychotics, antihistamines). Precipitating triggers set episodes in motion. Sleepwalkers may have subtle OSA or RLS, which can increase sleep pressure (priming) and sleep fragmentation (precipitating). Other sleepwalking triggers include stress, anxiety, migraine, fever, gastroesophageal reflux, excessive caffeine intake, a full bladder, and stimuli such as noise, light, or contact with a sleeping patient. Polysomnography is required if coexisting OSA is suspected, if events are frequent or stereotyped and seizures are suspected, or if patients do not respond to treatment or are injured.
Seizure disorders must also be considered with nocturnal motor behavior, especially when behaviors do not quite fit the profile of typical REM and NREM parasomnias. Seizure behaviors are usually stereotypical and repetitive, and patients often have a history of daytime epilepsy. When seizures are part of the differential diagnosis, patients require nocturnal polysomnography with a seizure montage.
Establishing safety for the sleepwalking patient is the first step in treatment. Precautions are described in Table 19-9. Most patients can be gently redirected back to bed, but touching or attempting to wake someone in the throes of an event may trigger an aggressive response. Patients need to be screened for sleep disorders that increase sleep pressure or disrupt sleep (e.g., OSA or RLS). They should stop taking possibly offending medications. Patients should be encouraged to improve sleep hygiene by keeping regular sleep schedules with adequate sleep opportunities. Sleep pressure can be decreased by extending sleep time even by 20-30 minutes per night to reduce sleep deprivation. Stress reduction at bedtime may diminish events. Relaxation techniques such as abdominal breathing, progressive relaxation, and self-hypnosis have been used with some success. Scheduled awakenings by parents before the child typically arouses can be helpful in some cases. For patients who do not respond to behavioral measures, low-dose clonazepam, TCAs, SSRIs, and melatonin have been found useful, although few studies support these treatments.
|
Table 19-9. Safety precautions for sleepwalkers |
||
|
||
Except for some skeletal muscles (e.g., eyes, diaphragm), we are paralyzed while we dream. Failure of muscle inhibition during REM sleep leads to acting out of dreams, sometimes with violent behaviors such as punching, kicking, and tackling that are likely to injure the patient or bed partner. REM sleep behavior disorder may herald neurodegenerative disease or be induced by commonly used medications (Arnulf 2012; Mahowald and Schenk 2004; Mahowald et al. 2011).
In contrast to sleepwalkers, whose eyes are open and whose arousal events typically occur in the first part of the sleep period, individuals with REM sleep behavior disorder enact their dreams with eyes closed during the second half of the sleep period, when REM sleep episodes increase in length and intensity. Whereas it is dangerous to attempt to wake a sleepwalker, waking a dreamer can avoid injury because patients with REM sleep behavior disorder wake fully alert and able to recount their dreams (unlike sleepwalkers, who might recall a snippet of a dream, if anything at all).
Loss of muscle atonia during REM sleep was previously thought to be rare and to occur primarily in men older than 50 years. Neurodegenerative diseases, such as Parkinson's disease, dementia with Lewy bodies, or multiple system atrophy, frequently coexist in older men with REM sleep behavior disorder, and REM sleep behavior disorder may precede their development, sometimes by many years. However, experts now suspect that REM sleep behavior disorder is more equally distributed in men and women and occurs in younger individuals more often than was previously reported, likely because of the widespread use of psychotropic medications. In people of all ages, REM sleep behavior disorder can be precipitated by antidepressants (TCAs, SSRIs, and monoamine oxidase inhibitors) as well as by alcohol consumption or excessive caffeine use. In addition, REM sleep behavior disorder coexists with narcolepsy in up to one-third of patients with that disease. When REM sleep behavior disorder occurs in young people or in women, the most likely causes are narcolepsy, medications, alcohol use, or excessive caffeine use.
Nocturnal in-lab polysomnography is needed to diagnose REM sleep behavior disorder. It will demonstrate increased muscle activity during REM sleep. Further evaluation of possibly inciting causes is also necessary.
To protect both the dreamer and his or her bed partner, the clinician should advise them to sleep in different rooms until treatment is effective. Nightly low-dose clonazepam (0.25 mg) reduces dream enactment but does not restore atonia. Melatonin has been found to increase REM sleep atonia.
Narcolepsy is a relatively uncommon cause of EDS, although its pathophysiology—the loss of sleep-state control—is reasonably well understood. The relevance of this disorder for psychiatric clinicians is that it coexists with a variety of disorders, including depression and anxiety. It also may present with symptoms of insomnia and sleep disruption. Some reports caution that the waking dreams of narcolepsy have led to an erroneous diagnosis of schizophrenia (Tsutsui et al. 2012; Walterfang et al. 2005).
Onset of narcolepsy is typically during adolescence, more rarely in childhood or later life. Narcolepsy can present with insomnia and sleep disruption, but EDS is usually the first symptom, followed (sometimes by several years) by episodes of cataplexy in about 70% of patients. Cataplexy is a sudden, short (from a few seconds to 2 minutes), sometimes almost imperceptible episode of lost muscle tone triggered by emotions, usually laughter, and may include slight facial weakness, grimaces, a head bob or jaw drop, knee weakness, blurred vision, slurred speech, or total collapse. Narcolepsy with cataplexy is associated with, low levels of cerebrospinal fluid hypocretin, typically less than one-third the levels found in healthy control subjects, or ≤110 pg/mL. Normal cerebrospinal fluid levels of hypocretin are present in patients who have narcolepsy without cataplexy (some of whom may experience "cataplexy-like" symptoms). Approximately 50% of patients in both groups (i.e., narcolepsy with and without cataplexy), as well as some patients without narcolepsy whose sleep is disrupted, report experiencing dreamlike imagery while falling asleep (hypnagogic hallucinations) or waking up (hypnopompic hallucinations) and sleep paralysis while falling asleep or waking due to REM sleep intrusion.
There are thought to be at least two forms of narcolepsy, which have been termed idiopathic narcolepsy and symptomatic narcolepsy (Nishino and Kanbayashi 2005). Idiopathic narcolepsy has long been known to have a genetic association with the human leukocyte antigen (HLA) HLA-DR2, further defined as primarily HLA-DQB1 *0602. This suggests a likely autoimmune etiology for at least some patients with narcolepsy who, for immunological reasons, lose hypothalamic neurons containing orexin/hypocretin and control of sleep switch mechanisms. Some cases of narcolepsy appear after infection (e.g., streptococcus, H1N1 swine flu) with possible immune system stimulation. There is a strong familial incidence of narcolepsy, as well as an association with the HLA system, which supports both a genetic and an immunological component to human narcolepsy. Intravenous immunoglobulin infusions have been reported to induce transient improvement in a few patients. Symptomatic narcolepsy, although rare, can be caused by other disturbances of brain or neurological function, including brain injury (Burgess and Scammell 2012).
Symptoms provide a high degree of diagnostic certainty for narcolepsy if cataplexy is clearly present; however, polysomnography is necessary to rule out other potential causes of EDS and hypersomnia. The nocturnal polysomnogram may show a sleep latency of less than 10 minutes and a REM sleep latency of less than 20 minutes. The multiple sleep latency test (four or five nap attempts following the nocturnal polysomnogram) usually demonstrates a sleep latency of 8 minutes or less and at least two sleep-onset REM sleep episodes in patients with narcolepsy. Patients should not be taking drugs that affect REM sleep or sleepiness for at least 14 days prior to testing. False-positive tests can occur in patients with circadian rhythm disorders, drug withdrawal, or OSA. Urine drug screening should precede the polysomnogram; the clinician should keep in mind that some individuals may feign symptoms of narcolepsy to obtain stimulant drugs.
Typically diagnosis and treatment of narcolepsy is by a sleep specialist or neurologist (Mignot 2012; Morgenthaler et al. 2007). Treatment is symptomatic because loss of hypocretin/orexin neurons cannot currently be reversed. Education, lifestyle changes, and medication can help alleviate symptoms in most cases and permit a relatively normal life. Treatment has the following components:
Once a sleep diagnosis (usually diagnoses) is suspected or identified, the nonspecialist clinician may choose to refer the patient to a sleep physician or clinic, proceed with further evaluation, or institute treatment him- or herself. Polysomnography is not necessary unless 1) sleep-related breathing disorder, narcolepsy, or REM sleep behavior disorder is suspected; 2) the causes of a patient's nocturnal movements require identification; or 3) insomnia treatment has been unsuccessful. A caveat: Although a polysomnogram should not replace the clinical evaluation, there are often surprises when one is obtained. Patients are frequently found to have more than one sleep disorder. An axiom of the sleep physician is that the longer we live, the longer the list of things that can disturb our sleep.
Disordered sleep imposes a heavy burden, especially for patients already weighed down by illness or stress; removal of this burden by identifying and treating the causes of sleep disturbance can make a tremendous difference in the lives of our patients.
Key Clinical Points
American Academy of Sleep Medicine: International Classification of Sleep Disorders, 2nd Edition: Diagnostic and Coding Manual. Westchester, IL, American Academy of Sleep Medicine, 2005
American Psychiatric Association: Diagnostic and Statistical Manual of Mental Disorders, 4th Edition. Washington, DC, American Psychiatric Association, 1994
American Psychiatric Association: Diagnostic and Statistical Manual of Mental Disorders, 4th Edition, Text Revision. Washington, DC, American Psychiatric Association, 2000
American Psychiatric Association: Diagnostic and Statistical Manual of Mental Disorders, 5th Edition. Arlington, VA, American Psychiatric Association, 2013
Arnulf I: REM sleep behavior disorder: motor manifestations and pathophysiology. Mov Disord 27:677-689, 2012
Bailey MJ, Coon SL, Carter DA, et al: Night/day changes in pineal expression of >600 genes: central role of adrenergic/cAMP signaling. J Biol Chem 284:7606-7622, 2009
Barkoukis TJ, Matheson JK, Ferber R, Doghramji K (eds): Therapy in Sleep Medicine. Philadelphia, PA, Elsevier, 2012
Beaudreau SA, Spira AP, Stewart A, et al: Validation of the Pittsburgh Sleep Quality Index and the Epworth Sleepiness Scale in older black and white women. Sleep Med 13:36-42, 2012
Besedovsky L, Lange T, Born J: Sleep and immune function. Pflugers Arch 463:121-137, 2012
Boksa P: Abnormal synaptic pruning in schizophrenia: urban myth or reality? J Psychiatry Neurosci 37:75-77, 2012
Bonnet MH, Arand DL: Hyperarousal and insomnia: state of the science. Sleep Med Rev 14:9-15, 2010
Bootzin RR, Epstein DR: Understanding and treating insomnia. Annu Rev Clin Psychol 7:435-58, 2011
Borbely AA, Achermann P: Sleep homeostasis and models of sleep regulation. J Biol Rhythms 14:557-568, 1999
Brostrom A, Sunnergren O, Johansson P, et al: Symptom profile of undiagnosed obstructive sleep apnoea in hypertensive outpatients in primary care: a structural equation model analysis. Qual Prim Care 20:287-298, 2012
Burgess CR, Scammell TE: Narcolepsy: neural mechanisms of sleepiness and cataplexy. J Neurosci 32:12305-12311, 2012
Buxton OM, Cain SW, O'Connor SP, et al: Adverse metabolic consequences in humans of prolonged sleep restriction combined with circadian disruption. Sci Transl Med 4:129-143, 2012
Cardinali DP, Furio AM, Reyes MP, et al: The use of chronobiotics in the resynchronization of the sleep-wake cycle. Cancer Causes Control 17:601-609, 2006
Carskadon MA, Vieira C, Acebo C: Association between puberty and delayed phase preference. Sleep 16:258-262, 1993
Donga E, van Dijk M, van Dijk JG, et al: A single night of partial sleep deprivation induces insulin resistance in multiple metabolic pathways in healthy subjects. J Clin Endocrinol Metab 95:2963-2968, 2010
Ebben MR, Spielman AJ: Non-pharmacological treatments for insomnia. J Behav Med 32:244-254, 2009
Erichsen D, Godoy C, Granse F, et al: Screening for sleep disorders in pediatric primary care: are we there yet? Clin Pediatr (Phila) 51:1125-1129, 2012
Feinberg I, Higgins LM, Khaw WY, et al: The adolescent decline of NREM delta, an indicator of brain maturation, is linked to age and sex but not to pubertal stage. Am J Physiol Regul Integr Comp Physiol 291:R1724-R1729, 2006
Fleetham J, Ayas N, Bradley D, et al: Canadian Thoracic Society 2011 guideline update: diagnosis and treatment of sleep disordered breathing. Can Respir J 18:25-47, 2011
Fort P, Bassetti CL, Luppi PH: Alternating vigilance states: new insights regarding neuronal networks and mechanisms. Eur J Neurosci 29:1741-1753, 2009
Franken P, Dijk DJ: Circadian clock genes and sleep homeostasis. Eur J Neurosci 29:1820-1829, 2009
Freeman ME, Kanyicska B, Lerant A, et al: Prolactin: structure, function, and regulation of secretion. Physiol Rev 80:1523-1631, 2000
Gaus SE, Strecker RE, Tate BA, et al: Ventrolateral preoptic nucleus contains sleep-active, galaninergic neurons in multiple mammalian species. Neuroscience 115: 285-294, 2002
Hofman MA, Swaab DF: The sexually dimorphic nucleus of the preoptic area in the human brain: a comparative morphometric study. J Anat 164:55-72, 1989
Huber R, Ghilardi MF, Massimini M, et al: Local sleep and learning. Nature 430:78-81, 2004
Ibrahim S, Foldvary-Schaefer N: Sleep disorders in pregnancy: implications, evaluation, and treatment. Neurol Clin 30:925-936, 2012
Innes KE, Selfe TK, Agarwal P: Prevalence of restless legs syndrome in North American and Western European populations: a systematic review. Sleep Med 12:623-634, 2011
Issa EB, Wang X: Sensory responses during sleep in primate primary and secondary auditory cortex. J Neurosci 28:14467-14480, 2008
Johns MW: A new method for measuring daytime sleepiness: the Epworth Sleepiness Scale. Sleep 14:540-545, 1991
Kadohira T, Kobayashi Y, Iwata Y, et al: Coronary artery endothelial dysfunction associated with sleep apnea. Angiology 62:397-400, 2011
Kasai T, Floras JS, Bradley TD: Sleep apnea and cardiovascular disease: a bidirectional relationship. Circulation 126:1495-1510, 2012
Kotagal S: Parasomnias in childhood. Sleep Med Rev 13:157-168, 2009
Kripke DF: An ultradian biologic rhythm associated with perceptual deprivation and REM sleep. Psychosom Med 34:221-234, 1972
Kryger MH, Roth T, Dement W: Principles and Practice of Sleep Medicine, 5th Edition. St. Louis, MO, Elsevier, 2011
Li AM, Au CT, Ho C, et al: Blood pressure is elevated in children with primary snoring. J Pediatr 155:362-368, 2009
Lockley SW, Evans EE, Scheer FA, et al: Short-wavelength sensitivity for the direct effects of light on alertness, vigilance, and the waking electroencephalogram in humans. Sleep 29:161-168, 2006
Lurie A: Endothelial dysfunction in adults with obstructive sleep apnea. Adv Cardiol 46:139-170, 2011
Luyster FS, Strollo PJ Jr, Zee PC, et al: Sleep: a health imperative. Sleep 35:727-734, 2012
Mahowald MW, Schenck CH: REM sleep without atonia: from cats to humans. Arch Ital Biol 142:469-478, 2004
Mahowald MW, Cramer Bornemann MA, Schenck CH: State dissociation, human behavior, and consciousness. Curr Top Med Chem 11:2392-2402, 2011
Marcus CL, Brooks LJ, Draper KA, et al: Diagnosis and management of childhood obstructive sleep apnea syndrome. Pediatrics 130:576-584, 2012a
Marcus CL, Brooks LJ, Draper KA, et al: Diagnosis and management of childhood obstructive sleep apnea syndrome. Pediatrics 130:e714-e755, 2012b
Matwiyoff G, Lee-Chiong T: Parasomnias: an overview. Indian J Med Res 131:333-337, 2010
Mignot EJ: A practical guide to the therapy of narcolepsy and hypersomnia syndromes. Neurotherapeutics 9:739-752, 2012
Morgenthaler TI, Lee-Chiong T, Alessi C, et al: Practice parameters for the clinical evaluation and treatment of circadian rhythm sleep disorders. An American Academy of Sleep Medicine report. Sleep 30:1445-1459, 2007
Morin CM, Benca R: Chronic insomnia. Lancet 379:1129-1141, 2012
Morin CM, Bootzin RR, Buysse DJ, et al: Psychological and behavioral treatment of insomnia: update of the recent evidence (1998-2004). Sleep 29:1398-1414, 2006
Naidoo N, Ferber M, Master M, et al: Aging impairs the unfolded protein response to sleep deprivation and leads to pro-apoptotic signaling. J Neurosci 28:6539-6548, 2008
National Center for Health Statistics: International Classification of Diseases, Ninth Revision, Clinical Modification (ICD-9-CM), 2011 Edition. Hyattsville, MD, National Center for Health Statistics, Centers for Disease Control and Prevention, 2011. Available at: http://www.cdc.gov/nchs/icd/icd9cm.htm. Accessed August 9, 2013.
Nishino S, Kanbayashi T: Symptomatic narcolepsy, cataplexy and hypersomnia, and their implications in the hypothalamic hypocretin/orexin system. Sleep Med Rev 9:269-310, 2005
Owens JA: Update in pediatric sleep medicine. Curr Opin Pulm Med 17:425—430, 2011
Perlis ML, Giles DE, Mendelson WB, et al: Psychophysiological insomnia: the behavioural model and a neurocognitive perspective. J Sleep Res 6:179-188, 1997
Pigeon WR, Pinquart M, Conner K: Metaanalysis of sleep disturbance and suicidal thoughts and behaviors. J Clin Psychiatry 73:ell60-ell67, 2012
Postuma RB, Aarsland D, Barone P, et al: Identifying prodromal Parkinson's disease: pre-motor disorders in Parkinson's disease. Mov Disord 27:617-626, 2012
Pressman MR: Factors that predispose, prime and precipitate NREM parasomnias in adults: clinical and forensic implications. Sleep Med Rev 11:5-30; discussion 31-33, 2007
Provini F, Tinuper P, Bisulli F, et al: Arousal disorders. Sleep Med 12 (suppl):S22-S26, 2011
Reid KJ, Zee PC: Circadian rhythm disorders. Semin Neurol 29:393-405, 2009
Reite M, Weissberg M, Ruddy J: Clinical Manual for the Evaluation and Treatment of Sleep Disorders. Washington, DC, American Psychiatric Publishing, 2009
Roenneberg X, Kuehnle T, Juda M, et al: Epidemiology of the human circadian clock. Sleep Med Rev 11:429-438, 2007
Rosenthal LD, Dolan DC: The Epworth Sleepiness Scale in the identification of obstructive sleep apnea. J Nerv Ment Dis 196:429-431, 2008
Roth T, Bogan RK, Culpepper L, et al: Excessive sleepiness: under-recognized and essential marker for sleep/wake disorder management. Curr Med Res Opin 26 (suppl):S3-S24; quiz S25-S27, 2010
Salas RE, Gamaldo CE, Allen RP: Update in restless legs syndrome. Curr Opin Neurol 23:401-106, 2010
Saper CB, Scammell TE, Lu J: Hypothalamic regulation of sleep and circadian rhythms. Nature 437:1257-1263, 2005
Saper CB, Fuller PM, Pedersen NP, et al: Sleep state switching. Neuron 68:1023-1042, 2010
Schaefer EW, Williams MV, Zee PC: Sleep and circadian misalignment for the hospitalist: a review. J Hosp Med 7:489-496, 2012
Schutte-Rodin S, Broch L, Buysse D, et al: Clinical guideline for the evaluation and management of chronic insomnia in adults. J Clin Sleep Med 4:487-504, 2008
Schwartz JR: Recognition of shift-work disorder in primary care. J Fam Pract 59:S18-S23, 2010
Senthilvel E, Auckley D, Dasarathy J: Evaluation of sleep disorders in the primary care setting: history taking compared to questionnaires. J Clin Sleep Med 7:41-48, 2011
Siebern AT, Manber R: New developments in cognitive behavioral therapy as the first-line treatment of insomnia. Psychol Res Behav Manag 4:21-28, 2011
Sieminski M: Restless legs syndrome: secondary, comorbid or coincidental? Eur Neurol 69:150-151, 2012
Sil A, Barr G: Assessment of predictive ability of Epworth scoring in screening of patients with sleep apnoea. J Laryngol Otol 126:372-379, 2012
Singletary KG, Naidoo N: Disease and degeneration of aging neural systems that integrate sleep drive and circadian oscillations. Front Neurol 2:66, 2011
Skene DJ, Arendt J: Circadian rhythm sleep disorders in the blind and their treatment with melatonin. Sleep Med 8:651-655, 2007
Slater G, Steier J: Excessive daytime sleepiness in sleep disorders. J Thorac Dis 4:608-616, 2012
Soreca I, Levenson J, Lotz M, et al: Sleep apnea risk and clinical correlates in patients with bipolar disorder. Bipolar Disord 14:672-676, 2012
Stenberg D: Neuroanatomy and neurochemistry of sleep. Cell Mol Life Sci 64:1187-1204, 2007
Tassinari CA, Rubboli G, Gardella E, et al: Central pattern generators for a common semiology in frontolimbic seizures and in parasomnias: a neuroethologic approach. Neurol Sci 26 (suppl):s225-s232, 2005
Tassinari CA, Cantalupo G, Hogl B, et al: Neuroethological approach to frontolimbic epileptic seizures and parasomnias: the same central pattern generators for the same behaviours. Rev Neurol (Paris) 165:762-768, 2009
Troxel WM, Buysse DJ, Matthews KA, et al: Sleep symptoms predict the development of the metabolic syndrome. Sleep 33:1633-1640, 2010
Tsutsui K, Kanbayashi T, Tanaka K, et al: Anti-NMDA-receptor antibody detected in encephalitis, schizophrenia, and narcolepsy with psychotic features. BMC Psychiatry 12:37, 2012
Viera AJ: Resistant hypertension. J Am Board Fam Med 25:487-495, 2012
Walterfang M, Upjohn E, Velakoulis D: Is schizophrenia associated with narcolepsy? Cogn Behav Neurol 18:113-118, 2005
Weber SA, Santos VJ, Semenzati Gde O, et al: Ambulatory blood pressure monitoring in children with obstructive sleep apnea and primary snoring. Int J Pediatr Otorhinolaryngol 76:787-790, 2012
Wells ME, Vaughn BV: Poor sleep challenging the health of a nation. Neurodiagn J 52:233-249, 2012
Wulff K, Porcheret K, Cussans E, et al: Sleep and circadian rhythm disturbances: multiple genes and multiple phenotypes. Curr Opin Genet Dev 19:237-246, 2009
Zadra A, Desautels A, Petit D, et al: Somnambulism: clinical aspects and pathophysiological hypotheses. Lancet Neurol 12:285-294, 2013
Barkoukis TJ, Matheson JK, Ferber R, Doghramji K (eds): Therapy in Sleep Medicine. Philadelphia, PA, Elsevier, 2012
Kryger MH, Roth T, Dement W: Principles and Practice of Sleep Medicine, 5th Edition. St. Louis, MO, Elsevier, 2011
Perlis ML, Aloia M, Kuhn B (eds): Behavioral Treatments for Sleep Disorders: A Comprehensive Primer of Behavioral Sleep Medicine Interventions (Practical Resources for the Mental Health Professional). London, Academic Press, 2011
Reite M, Weissberg M, Ruddy J: Clinical Manual for the Evaluation and Treatment of Sleep Disorders. Washington, DC, American Psychiatric Publishing, 2009
American Academy of Sleep Medicine practice guidelines (http://www.aasmnet.org/practiceguidelines.aspx)
Appendix: DSM-5 Diagnostic Criteria for Selected Common Sleep Disorders
|
Box 19-1. DSM-5 Criteria for Circadian Rhythm Sleep-Wake Disorders |
Specify whether: Delayed sleep phase type Specify if: Familial Specify if: Overlapping with non-24-hour sleep-wake type Advanced sleep phase type Specify if: Familial Irregular sleep-wake type Non-24-hour sleep-wake type Shift work type Unspecified type Specify if: Episodic Persistent Recurrent |
NOTICE. Criteria set above contains only the diagnostic criteria and specifiers; refer to DSM-5 for the full criteria set, including specifier descriptions and coding and reporting procedures.
|
Box 19-2. DSM-5 Criteria for Obstructive Sleep Apnea Hypopnea |
Specify current severity: Mild Moderate Severe |
NOTICE. Criteria set above contains only the diagnostic criteria and specifiers; refer to DSM-5 for the full criteria set, including specifier descriptions and coding and reporting procedures.
|
Box 19-3. DSM-5 Criteria for Insomnia Disorder |
Specify if: With non-sleep disorder mental comorbidity With other medical comorbidity With other sleep disorder Specify if: Episodic Persistent Recurrent Note: Acute and short-term insomnia (i.e., symptoms lasting less than 3 months but otherwise meeting all criteria with regard to frequency, intensity, distress, and/or impairment) should be coded as an other specified insomnia disorder. |
NOTICE. Criteria set above contains only the diagnostic criteria and specifiers; refer to DSM-5 for the full criteria set, including specifier descriptions and coding and reporting procedures.
|
Box 19-4. DSM-5 Criteria for Restless Legs Syndrome |
|
NOTICE. Criteria set above contains only the diagnostic criteria and specifiers; refer to DSM-5 for the full criteria set, including specifier descriptions and coding and reporting procedures.
|
Box 19-5. DSM-5 Criteria for Non-Rapid Eye Movement Sleep Arousal Disorders |
Specify whether: Sleepwalking type Specify if: With sleep-related eating With sleep-related sexual behavior (sexsomnia) Sleep terror type |
NOTICE. Criteria set above contains only the diagnostic criteria and specifiers; refer to DSM-5 for the full criteria set, including specifier descriptions and coding and reporting procedures.
|
Box 19-6. DSM-5 Criteria for Rapid Eye Movement Sleep Behavior Disorder |
|
NOTICE. Criteria set above contains only the diagnostic criteria and specifiers; refer to DSM-5 for the full criteria set, including specifier descriptions and coding and reporting procedures.
|
Box 19-7. DSM-5 Criteria for Narcolepsy |
Specify whether: Narcolepsy without cataplexy but with hypocretin deficiency Narcolepsy with cataplexy but without hypocretin deficiency Autosomal dominant cerebellar ataxia, deafness, and narcolepsy Autosomal dominant narcolepsy, obesity, and type 2 diabetes Narcolepsy secondary to another medical condition Specify current severity: Mild Moderate Severe |
NOTICE. Criteria set above contains only the diagnostic criteria and specifiers; refer to DSM-5 for the full criteria set, including specifier descriptions and coding and reporting procedures.